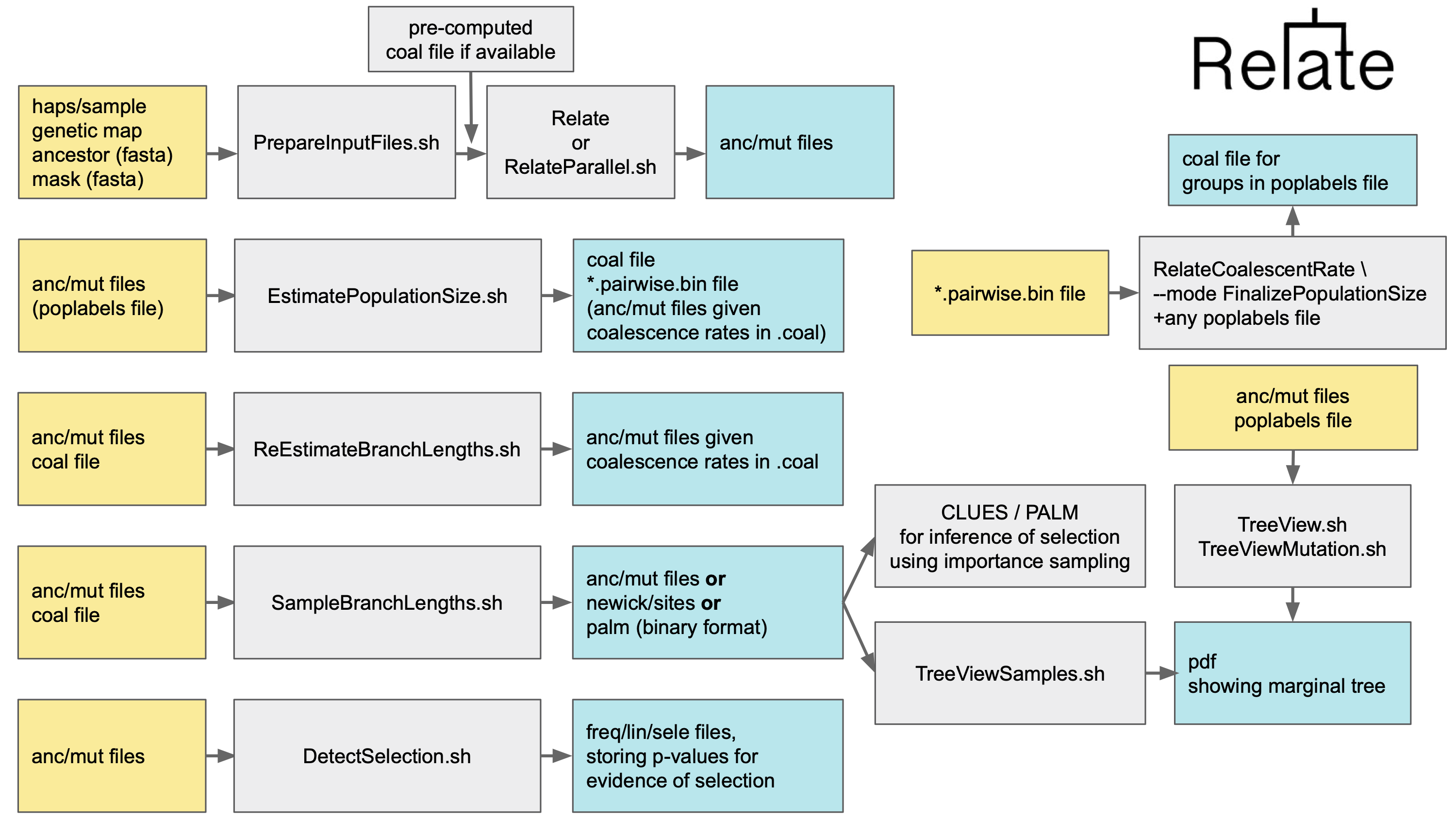
Software to estimate genome-wide genealogies for thousands of samples
Relate estimates genome-wide genealogies in the form of trees that adapt to changes in local ancestry caused by recombination. The method, which is scalable to thousands of samples, is described in the following paper. Please cite this paper if you use our software in your study.
Citations:
- (original Relate paper) Leo Speidel, Marie Forest, Sinan Shi, Simon Myers.
A method for estimating genome-wide genealogies for thousands of samples. Nature Genetics 51: 1321-1329, 2019. - (update, v1.1.*) Leo Speidel, Lara Cassidy, Robert W. Davies, Garrett Hellenthal, Pontus Skoglund, Simon R. Myers.
Inferring population histories for ancient genomes using genome-wide genealogies. Molecular Biology and Evolution 38: 3497–3511, 2021.
Website: https://leospeidel.com
Download
Relate is available for academic use. To see rules for non-academic use, please read the LICENCE file, which is included with each software download.
Pre-compiled binaries (last updated: 13/09/2025)
Github repository
Alternatively, you can compile your own version by downloading the source code from this github repository.
In the downloaded directory, we have included a toy data set. You can try out Relate using this toy data set by following the instructions on our getting started page.
If you have any problems getting the program to work on your machine or would like to request an executable for a platform not shown here, please send a message to leo.speidel [at] outlook [dot] com.
We document changes to previous versions in a change-log.
What's new?
We provide ancestral genomes, genomic mask files, precomputed coalescence rates, and recombination rates for hg37 and hg38 here.
Versions >1.1.0 include a few new features such as:- Allowing for non-contemporary samples, e.g., ancient DNA: Simply add the --sample_ages option.
- Allowing for haploid organisms: set the ID_2 column in the sample file to NA and Relate will work for haploid organisms.
- Improved coalescence rate estimation: The algorithm has been improved and behaviour has changed slightly. The output now always contains all trees, and the threshold parameter only affects iterations leading up to the last one. The threshold parameter now is a float in [0,1], specifying the proportion of trees to throw away (Default = 0.5).
- Arbitrary strings as chromosome IDs: in scripts such as EstimatePopulationSize.sh, use the --chr option to specify a file containing chromosome name.
- More flexible epochs for coalescence rate estimation: use the option --bins to specify epoch boundaries in the format 10^seq(x,y,stepsize).
1000 Genomes coalescence rates, allele ages, and selection p-values
We deposited Relate-estimated coalescence rates, allele ages, and p-values for evidence of positive selection for the 1000 Genomes Project here.
These were obtained by estimating the joint genealogy of all 1000 GP populations and then extracted the embedded genealogy for each population.
For the genealogy of each population, we jointly estimated the population size history and branch lengths.
Variants segregating in more than one population therefore have correlated but different allele ages in each population.
Inferred trees
You can download inferred trees from the links below:
Relater: R package for handling Relate output files
I made an R package for handling Relate output files (MIT license). It's still under development but the main functionality is working.
You can install this package, e.g., usinglibrary(devtools)
devtools::install_github("leospeidel/relater")relate_lib: Conversion to tskit format
This github repo contains functions to convert between Relate and tskit format (MIT license).
It doubles as a C++ library you can link to if you want to use some of our Relate functions in C++.
relate_lib/bin/Convert --mode ConvertFromTreeSequence \
--anc example.anc.gz \
--mut example.mut.gz \
-i examplerelate_lib/bin/Convert --mode ConvertToTreeSequence \
--anc example.anc.gz \
--mut example.mut.gz \
-o exampleThanks to Nathaniel S. Pope, you can now also specify an argument that compresses these Relate-converted tree sequences by assigning the same age to nodes with identical descendant sets across adjacent trees.
relate_lib/bin/Convert --mode ConvertToTreeSequence \
--compress \
--anc example.anc.gz \
--mut example.mut.gz \
-o exampleAdd-on modules
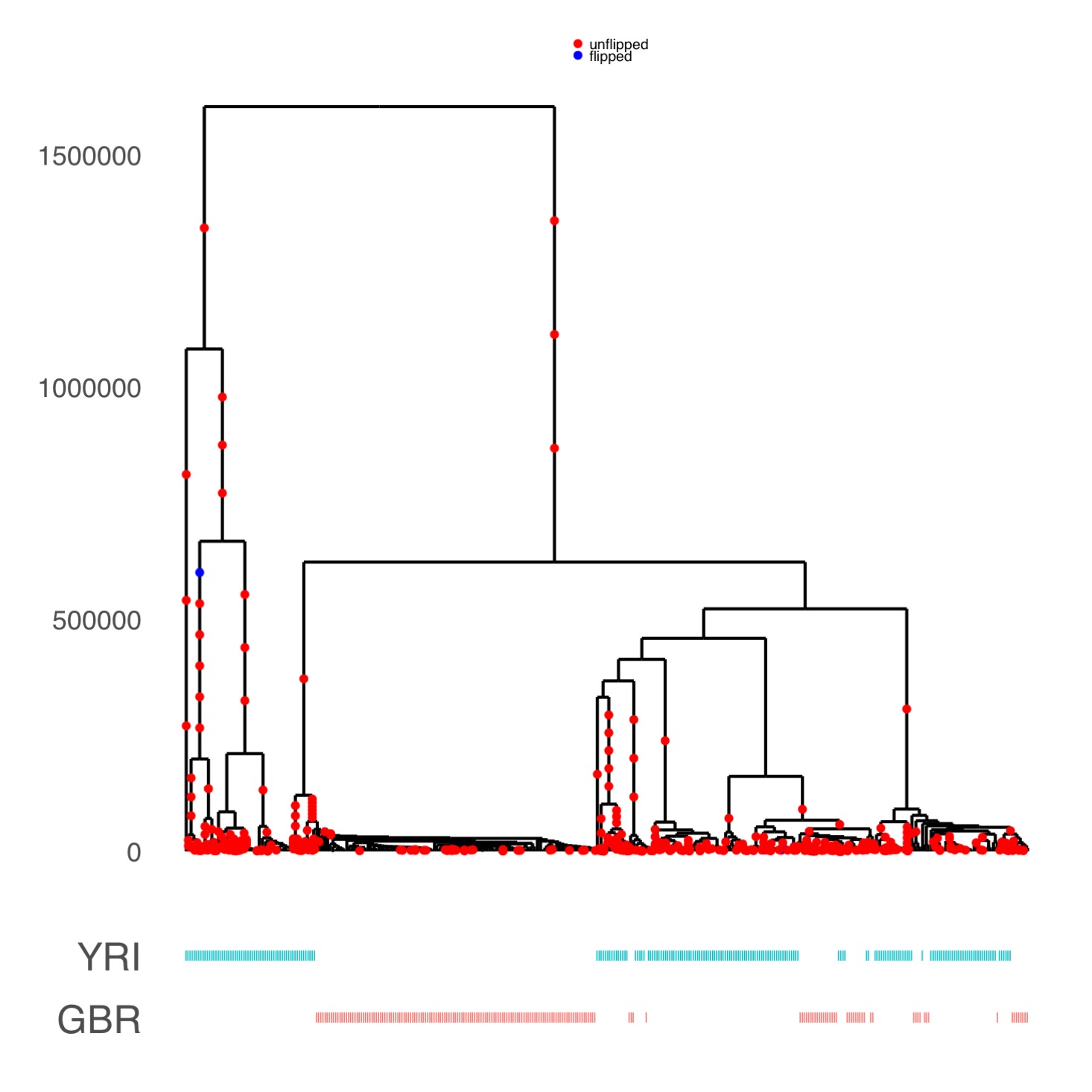 Relate can be used to plot a marginal tree of interest. This tree corresponds to the LCT region, where a mutation at SNP rs4988235 is believed to be responsible for Lactose tolerance in Europeans (here GBR). We can see that the derived allele at this SNP has spread rapidly in GBR, which is indicative of strong positive selection.
Relate can be used to plot a marginal tree of interest. This tree corresponds to the LCT region, where a mutation at SNP rs4988235 is believed to be responsible for Lactose tolerance in Europeans (here GBR). We can see that the derived allele at this SNP has spread rapidly in GBR, which is indicative of strong positive selection. 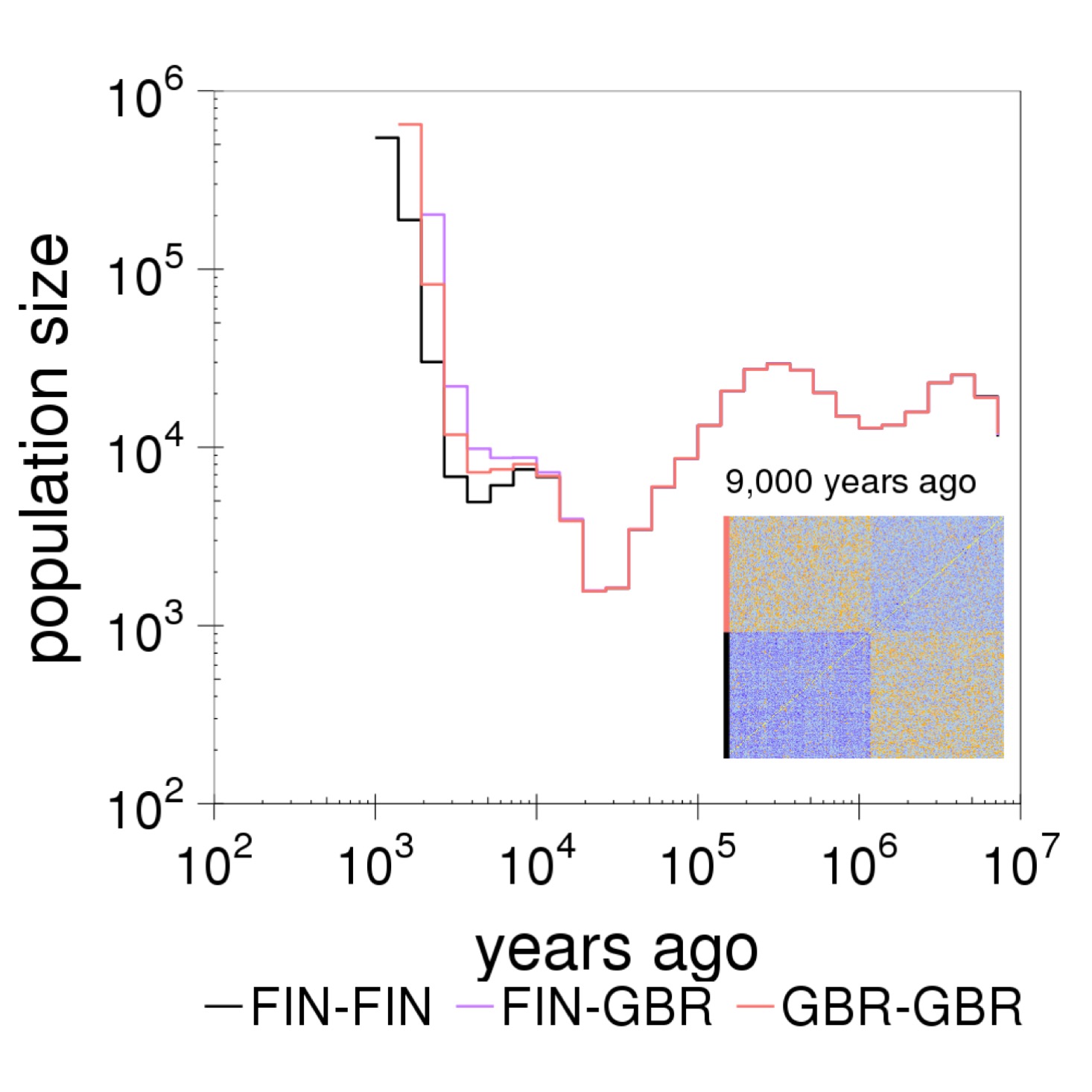 Relate can be used to estimate population sizes and separation histories between populations. The figure shows the separation history between FIN and GBR in the 1000GP data set. The inset shows the matrix of coalescent rates between pairs of haplotypes 9,000 years ago. Rows and columns are sorted by population labels of haplotypes, as indicated by the colour on the left of the matrix.
Relate can be used to estimate population sizes and separation histories between populations. The figure shows the separation history between FIN and GBR in the 1000GP data set. The inset shows the matrix of coalescent rates between pairs of haplotypes 9,000 years ago. Rows and columns are sorted by population labels of haplotypes, as indicated by the colour on the left of the matrix. 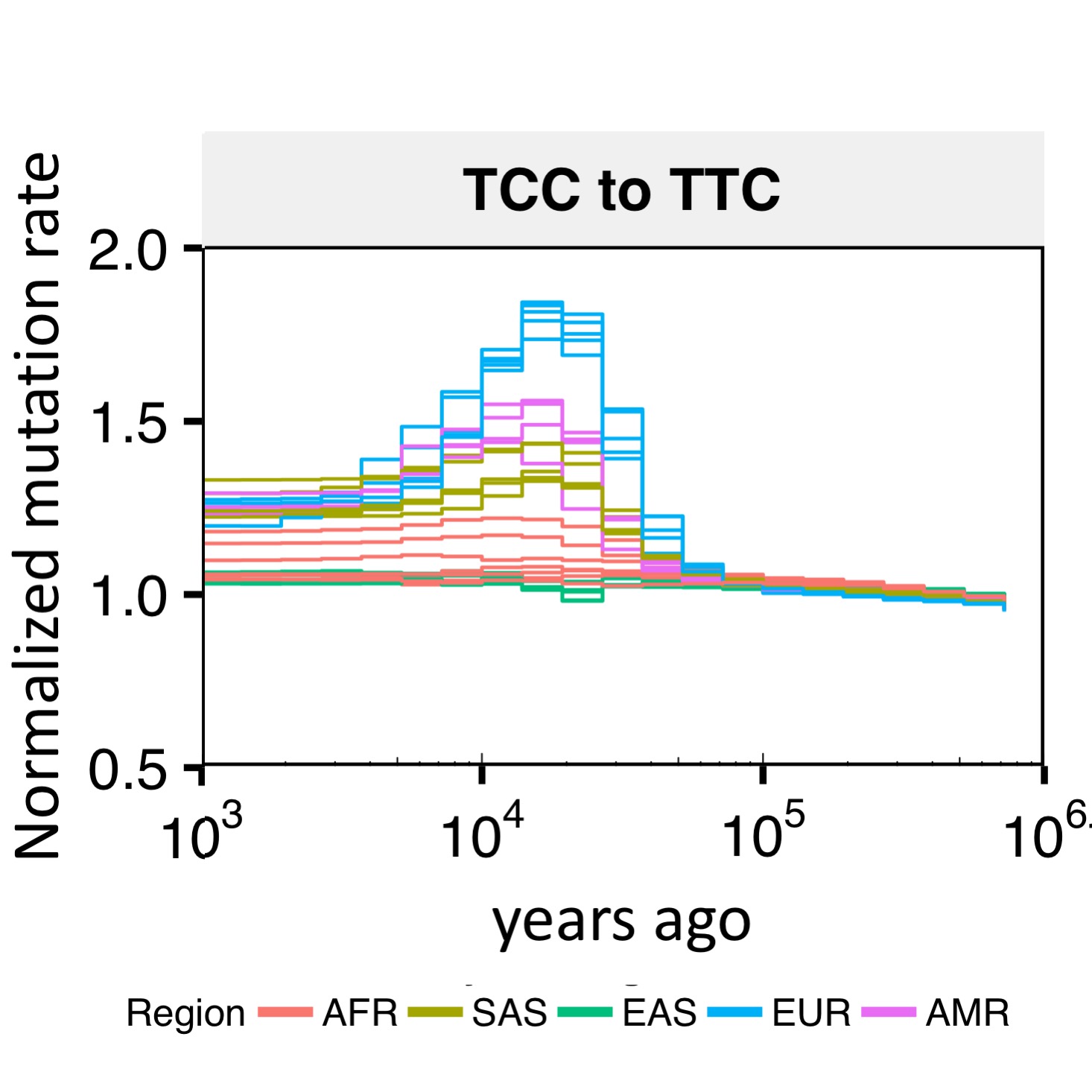 Relate can be used to estimate mutation rates through time. The figure shows TCC to TTC mutation rates for all 26 populations in the 1000GP data set. Trends shared between mutation categories are eliminated by dividing by the overall average mutation rate. For each population, the mutation rate is normalized such that its mean over time equals one. Consistent with previous estimates, we see an increase in the mutation rate of TCC to TTC mutations about 10,000 to 30,000 years ago in Europeans and Southern Asians.
Relate can be used to estimate mutation rates through time. The figure shows TCC to TTC mutation rates for all 26 populations in the 1000GP data set. Trends shared between mutation categories are eliminated by dividing by the overall average mutation rate. For each population, the mutation rate is normalized such that its mean over time equals one. Consistent with previous estimates, we see an increase in the mutation rate of TCC to TTC mutations about 10,000 to 30,000 years ago in Europeans and Southern Asians.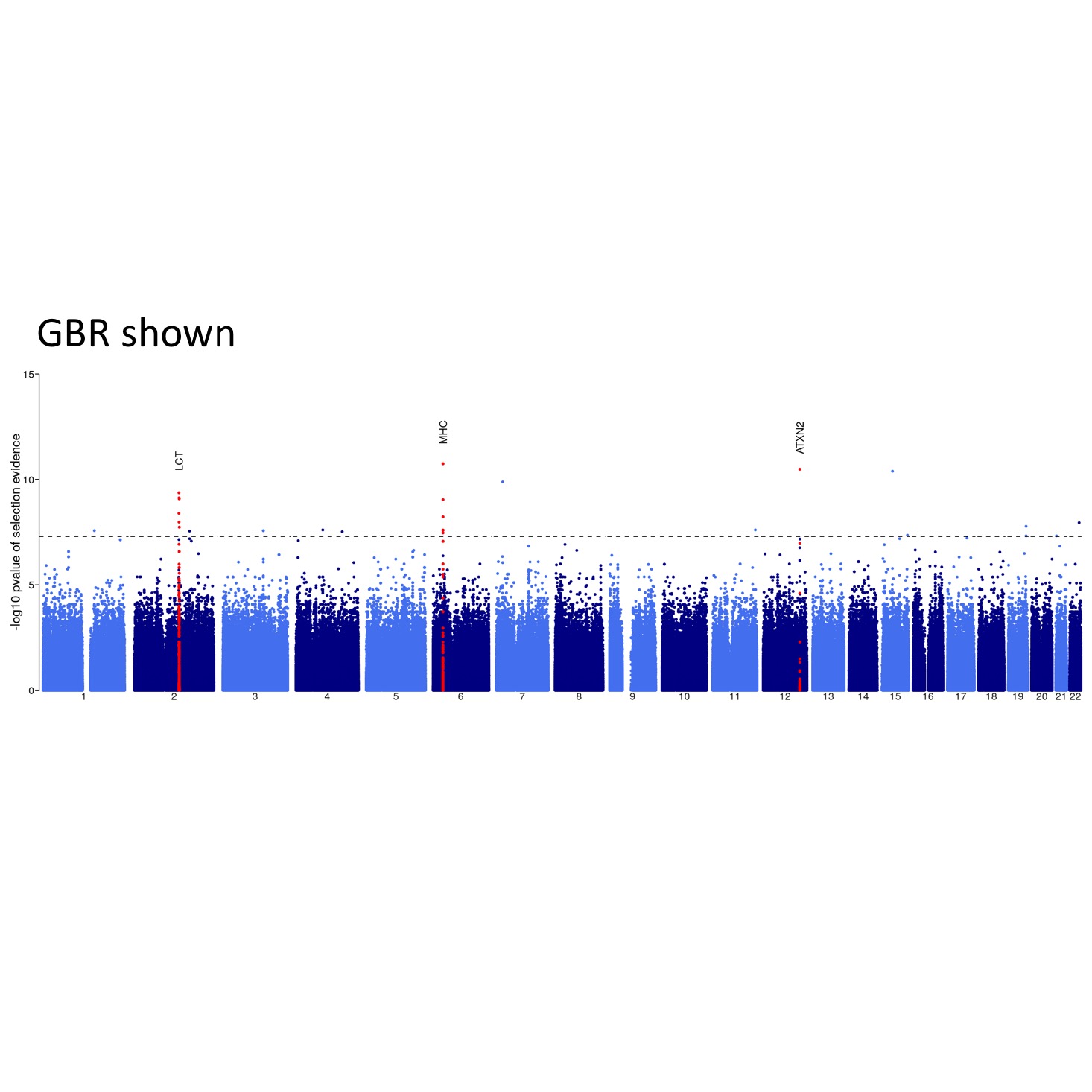 Relate can be used to detect evidence for positive selection. We calculate a p-value for selection evidence that quantifies how quickly a mutation has spread in the population. The figure shows a manhattan plot for GBR which indicates clear peaks in the LCT and MHC regions, which are known targets of positive selection.
Relate can be used to detect evidence for positive selection. We calculate a p-value for selection evidence that quantifies how quickly a mutation has spread in the population. The figure shows a manhattan plot for GBR which indicates clear peaks in the LCT and MHC regions, which are known targets of positive selection.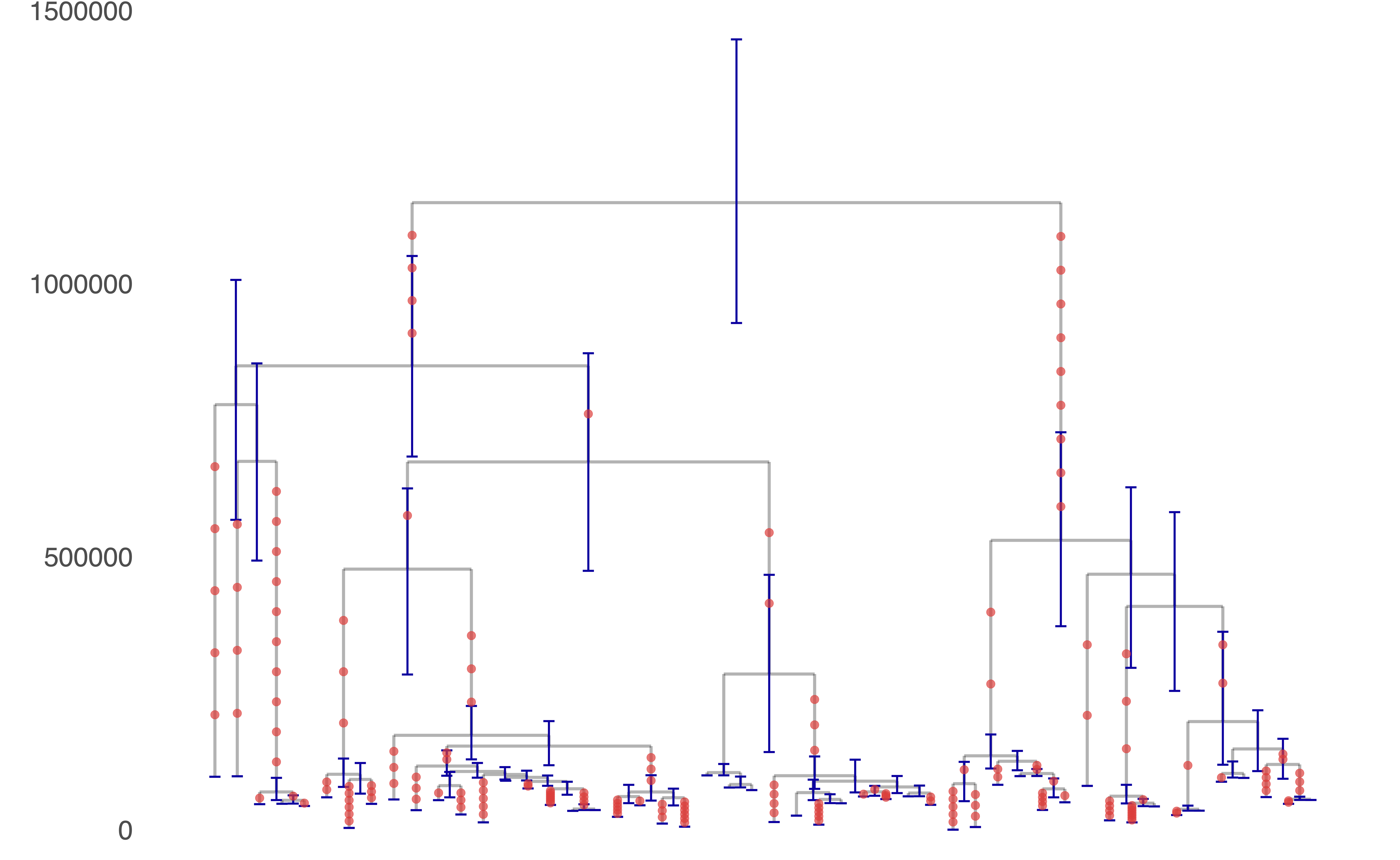 Relate can infer genealogies for non-contemporary samples, such as high coverage ancient genomes or time-stamped samples of bacteria or viruses. This plot was generated using the TreeViewSamples.sh script, using 100 sampled branch lengths. The tree represents the posterior mean times, and "error bars" indicate the 0.025 and 0.975 quantiles of the posterior density of coalescence ages.
Relate can infer genealogies for non-contemporary samples, such as high coverage ancient genomes or time-stamped samples of bacteria or viruses. This plot was generated using the TreeViewSamples.sh script, using 100 sampled branch lengths. The tree represents the posterior mean times, and "error bars" indicate the 0.025 and 0.975 quantiles of the posterior density of coalescence ages.
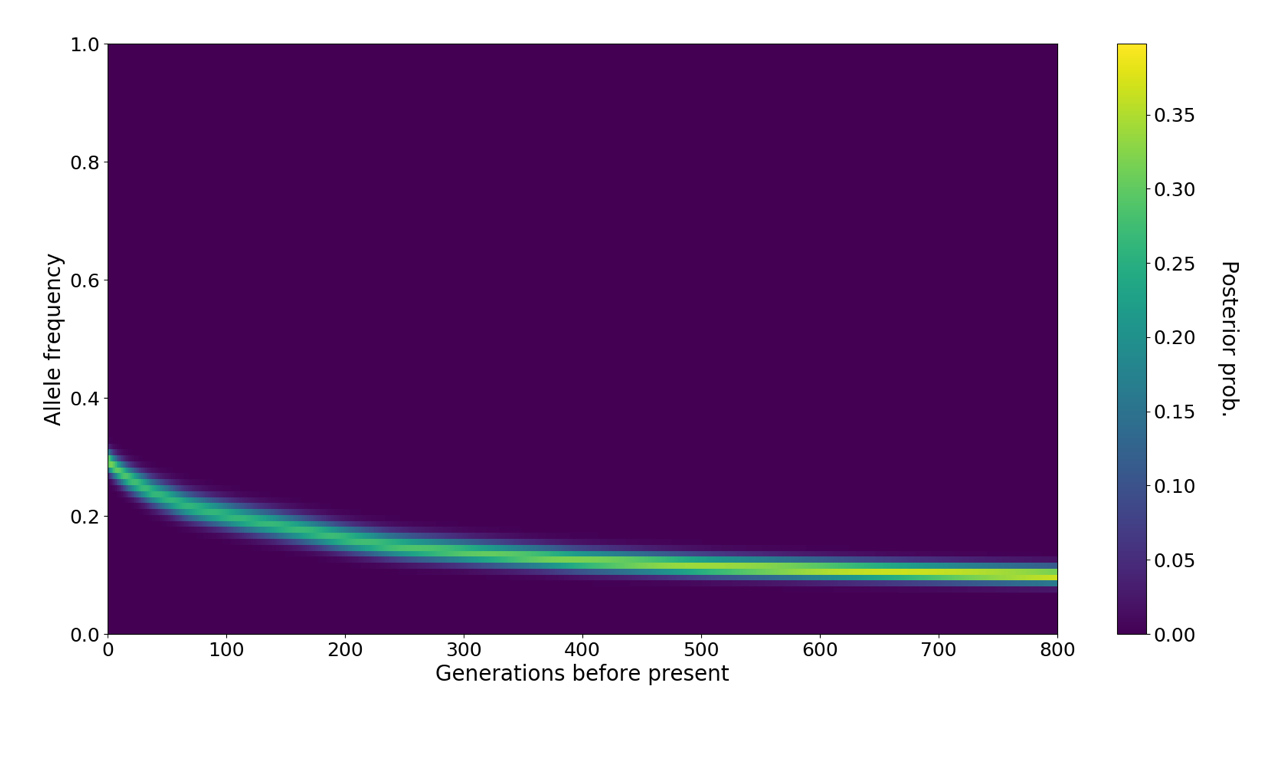 Using the SampleBranchLengths.sh script, Relate can sample branch lengths from the posterior, which can then be used e.g., by CLUES to infer allele frequency trajectories and selection coefficients using an importance sampling scheme. See the CLUES repo for more details.
Using the SampleBranchLengths.sh script, Relate can sample branch lengths from the posterior, which can then be used e.g., by CLUES to infer allele frequency trajectories and selection coefficients using an importance sampling scheme. See the CLUES repo for more details.
Change-log
- Updated interpretation of rho in --painting. Using 2500*20/N*rho.
- Reimplemented OptimizeParameters
- Updated RelateCoalescentRate to average better over ancient DNA time epochs
- Removed focal SNP weighting in BuildTopology
- Bug fix introduced in v1.2.1 which led to execution errors when running RelateSelection
- Bug fix introduced in v1.2.0 which led to bad topologies for larger sample sizes
- Bug fix introduced in v1.2.0 in EstimatePopulationSize.sh which caused an error when using with multiple chromosomes
- Increased consistency between adjacent trees and improved memory footprint.
- Added option to force build trees every x bases.
- RelateSelection --mode Frequency works now with ancient genomes.
v1.1.*
Pre-compiled binaries (last updated: 14/06/2022)
- Fixed bug in TreeView where singletons were not plotted.
- Made ConvertToTreeSequence consistent with relate_lib version.
- Bug fix in SummarizeMutationRates (assumed float instead of double, leading to Segfault).
- Updated initialisation of bl with ancients.
- Small bug fixes.
- Added option to exclude transitions in branch length estimation (useful for ancient genomes).
- Modified tree builder when ancients samples are specified (see Colate paper for details).
- Fixed a in ConvertFromVcf that caused empty haps/sample files to be outputted
- Added a function in RelateExtract to get branch frequencies for every SNP.
- Fixed a major bug (v1.1.* onwards) in EstimatePopulationSize.sh when setting seed (not affected when seed is not set).
- Added RelateSlurm.sh and RelateLSF.sh
- Added a function to retrospectively map a mutation to trees in RelateExtract
- Fixed bug in PrepareInputFiles, when downsampling individuals (introduced in v1.1.0).
- Changed parameters for branch lengths estimation for aDNA, to improve mixing properties of MCMC.
- Changed function for anc to newick conversion, to allow multiple trees to be extracted.
- Changed filename used for CLUES and PALM from *.palm to *.timeb.
- Fixed bug in EstimatePopulationSize.sh
- Allowing for non-contemporary samples, e.g., ancient DNA: Simply add the --sample_ages option.
- Allowing for haploid organisms: set the ID_2 column in the sample file to NA and Relate will work for haploid organisms.
- Improved coalescence rate estimation: The algorithm has been improved and behaviour has changed slightly. The output now always contains all trees, and the threshold parameter only affects iterations leading up to the last one. The threshold parameter now is a float in [0,1], specifying the proportion of trees to throw away (Default = 0.5).
- Arbitrary strings as chromosome IDs: in scripts such as EstimatePopulationSize.sh, use the --chr option to specify a file containing chromosome name.
- More flexible epochs for coalescence rate estimation: use the option --bins to specify epoch boundaries in the format 10^seq(x,y,stepsize).
v1.0.*
Pre-compiled binaries (last updated: 10/01/2020)
- Input files can now have CHR, RSID, ANC, DER columns of type strings (up to 1024 chars).
- Added a scripts named TreeViewMutation.sh, taking same input as TreeView.sh, but different visualisation (bp_of_interest has to be SNP).
- Fixed a bug in ConvertToTreeSequence that meant internal nodes were flagged as sample nodes.
- Fixed a small bug that meant memory was misallocated when the --memory value was set too large.
- Fixed a bug in PrepareInputFiles.sh that meant the script terminated before filtering SNPs using a genomic mask when input was not gzipped.
- Changed the default for y axis scale in TreeView to not be a log-scale.
- Modified default criteria to terminate population size estimation. Now terminating when iteration at least 2 and "mean absolute error"/mu less than 0.1.
- Modified memory allocation in Relate, where we were allocating too much memory for very small sample sizes (N less than 10).
- Modified error message when poplabels file is misspecified in FinalizePopulationSize - you can just rerun this step again at the end.
- Included the script RelateSGE.sh for running Relate on a SGE cluster.
- Fixed a bug in FinalizePopulationSize --mode EstimatePopulationSize, where I had forgotten to close a file leading to an error on some platforms.
- Allowing .poplabels file to be tab separated.
- Fixed a bug in TreeView which threw an error when completely fixed SNPs were included in the haps/sample file.
- Fixed a bug in FinalizePopulationSize introduced in v1.0.9, which threw an error message for correct input files.
- Fixed a bug in SampleBranchLengths which was introducing small rounding errors.
- Fixed a bug in SampleBranchLengths.sh, where a filename was hard-coded by mistake.
- Implemented a module which samples branch lengths from the posterior given tree topologies and effective population size histories.
- Added some more detail to documentation for RelateSelection.
- Added a more meaningful error message when .poplabels file is misspecified in EstimatePopulationSize.sh.
- Now requiring different input and output names for DetectSelection.sh when it overwrites files otherwise.
- DetectSelection.sh now works also if additional columns are not appended to the .mut file using ./RelateFileFormats --mode GenerateSNPAnnotations. However, code is more efficient if these columns are appended.
- Switched to using R package cowplot in TreeView.sh.
- Fixed bug in the painting which affects tree topologies sometimes, but has only a small effect (not visible in distance measures when comparing to truth in simulations).
- I added a function for converting anc/mut output files to tree sequence file format (tskit). Currenty, some information is lost by this conversion.
- I had introduced a bug in update v1.0.4 in the function RelateExtract --mode TreeAtSNPAsNewick which has been fixed.
- Substituted zcat with gunzip -c to fix a bug in Macs.
- Added requirement of R version >= 3.3.1 for TreeView because of a known bug with grid.draw() in older versions.
- Fixed bug in RelateExtract --mode TreeAtSNPAsNewick, which outputted the first tree when snp_of_interest was not a SNP in the data set. Changed option snp_of_interest to pos_of_interest and this function now prints the tree at the position of interest. In addition, output filename was not using the option -o, which has been corrected.
- Implemented pipeline for calculating p-values for selection evidence. Updated corresponing entry in the documentation.
- Bug fix in RelateSelection --mode Frequency: Previous version had a bug whenever two internal nodes had same age.
- Fixed function RelateFileFormats --mode ConvertFromVcf which previously parsed the vcf incorrectly in some cases.
- Implemented parallelization of module EstimatePopulationSize.
- Bug fix in parsing function of haps/sample.
- Initial release.