Overview
This page lists add-on modules that can be applied to the Relate output for inference of coalescnece rates, mutation rates, and selection.
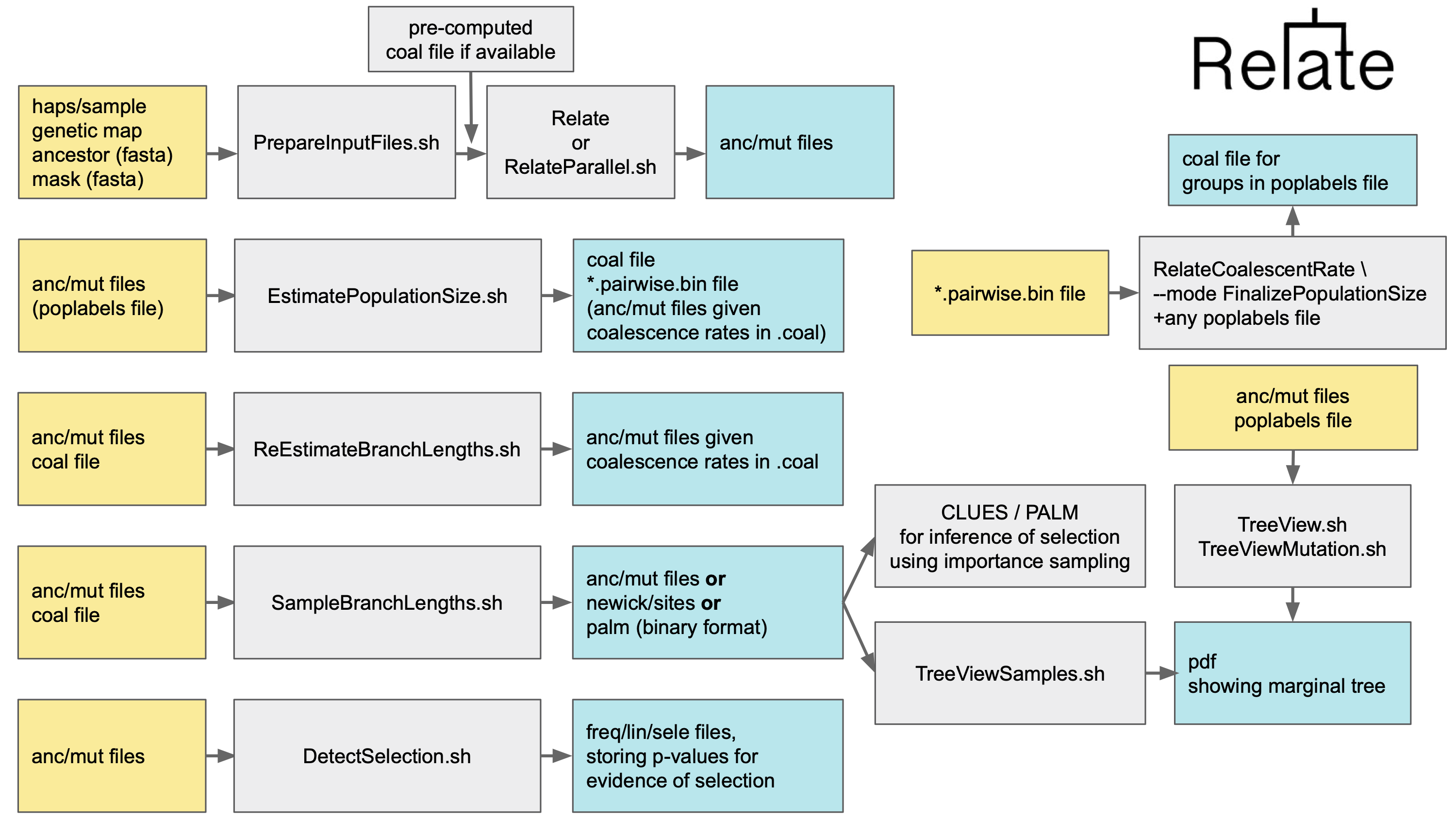
Estimate coalescence rates and effective population sizes
Estimating effective population sizes
You can find a bash script under
PATH_TO_RELATE/scripts/EstimatePopulationSize/EstimatePopulationSize.sh- estimates population sizes
- re-estimates branch lengths using the estimated population sizes
- estimates an average mutation rate.
Please note that two versions of coalescence rates/population sizes are outputted:
- output.coal: Treats all samples as one group.
- *.pairwise.coal/.bin, reflects grouping in poplabels file. You can rerun RelateCoalescentRates --mode FinalizePopulationSize to extract coalescence rates for any .poplabels file.
You can test the code on the toy data set in example/data.
Do not move the script - we are extracting the path to the binaries from the location of the script!
PATH_TO_RELATE/scripts/EstimatePopulationSize/EstimatePopulationSize.sh \
-i example \
-m 1.25e-8 \
--poplabels data/example.poplabels \
--seed 1 \
-o example_popsizeInput arguments
- -i,--input
- Filename of the .anc and .mut files, excluding the file extensions.
- -o,--output
- Filename of output files without file extensions.
- -m,--mutation_rate
- Mutation rate per base per generation.
- --poplabels
- Filename of a .poplabels file. The file format is described here. This file needs to always contain all samples.
- --noanc
- Optional: Flag specifying whether to reinfer branch lengths at the end. 0: reinfer, 1: don't reinfer. Default: 0.
- --pop_of_interest
- Optional: Specifies populations for which population size is estimated. The second column of the .poplabels file is used for population labels. You can specify multiple populations separated by commas, e.g. pop1,pop2. When using this option, it is recommended (but NOT necessary), to run Relate with --annot. This can also be done after running Relate without --annot, see here.
- --first_chr & --last_chr
- Optional: Allow for multiple input files, where filenames are assumed to be numbered by chromosome index.
Example: -i example --first_chr 1 --last_chr 2 assumes example_chr1.anc, example_chr1.mut, example_chr2.anc, example_chr2.mut as input. - --chr
- Optional: File containing chromosome names (one per line). Overrides --first_chr and --last_chr.
Example: -i example --chr chr.txt assumes example_chr*.anc, example_chr*.mut as input. - --threads
- Optional: Maximum number of threads. Default is 1.
- --bins
- Optional: Specify epoch bins. Format: lower, upper, stepsize for function c(0,10^seq(lower, upper, stepsize)).
- --years_per_gen
- Optional: Years per generation. Default: 28.0
- --num_iter
- Optional: Number of iterations of the algorithm. Default is 5.
- --threshold
- Optional: Float in [0,1] which is the fraction of trees to be dropped, ranked by number of mutations mapping. This option serves two purposes: First, it removes bad/uninformative trees. Second it speeds up the inference. As we increase this value, we remove more trees. Default: 0.5.
- --seed
- Optional: Seed for the random number generator used in the MCMC algorithm for estimating branch lengths.
- --noplot
- Optional: Takes no value. If specified, do not plot the results using R.
Output
| example_popsize.pdf | A plot showing the estimated population size. |
| example_popsize.anc.gz, example_popsize.mut.gz, example_popsize.dist | Not outputted if --noanc 1. Genealogy with updated branch lengths. Will contain all trees regardless of threshold value. If --noanc 1 is specified, you can separately reestimate branch lengths using the ReEstimateBranchLengths.sh script. |
| example_popsize.coal | Contains coalescence rates and cross-coalescence rates, treating all samples as one population (see below for file format). |
| example_popsize.pairwise.coal and *.bin | Coalescence rate file and corresponding binary file containing coalescence rates between pairs of samples. Can be used by RelateCoalescentRates --mode FinalizePopulationSize to extract coalescence rates for any .poplabels file. |
| example_popsize_avg.rate | Contains average mutation rate through time. Please note that any trends in the average mutation rate are confounded by coalescence rate changes - Relate can only infer mutation rate changes in mutation subcategories (see estimate mutation rate section). |
File formats
Reestimate branch lengths
Reestimate branch lengths using MCMC given coal file.
For a given coal file, this script will output anc/mut files with posterior mean branch lengths given coalescence rates.
- -i,--input
- Filename of .anc and .mut files without file extension. Needs to contain all SNPs in input haps/sample files. If not, see --dist option.
- -o,--output
- File of the output files without file extensions.
- -m,--mu
- Mutation rate, e.g., 1.25e-8.
- --coal
- Estimated coalescent rate, treating all haplotypes as one population.
- --threads
- Optional: Maximum number of threads. Default is 1.
- --first_bp
- Optional: First bp position.
- --last_bp
- Optional: Last bp position.
- --dist
- Optional: Filename containing bp positions of SNPs and distances between adjacent SNPs (file format). E.g., obtained using RelateExtract --mode AncMutForSubregion. Required if anc/mut file only covers a subregion of input haps/sample files.
- --seed
- Optional: Random seed for branch lengths estimation.
PATH_TO_RELATE/scripts/SampleBranchLengths/ReEstimateBranchLengths.sh \
-i example \
-o example_sub \
-m 1.25e-8 \
--coal popsize.coal \
--first_bp 100000 \
--last_bp 200000 \
--seed 1 More details
Below, we describe details about the functions used in the script.- Estimate average mutation rate
- Estimate coalescence rate
- Re-estimate branch lengths
Calculate coalescence rates through time
- -i,--input
- Filename of the .anc and .mut files, excluding the file extensions.
- -o,--output
- Filename of output files without file extensions.
- -m,--mutation_rate
- Mutation rate per base per generation.
- --poplabels
- Optional: Filename of a .poplabels file. The file format is described here.
- --first_chr & --last_chr
- Optional: Allow for multiple input files, where filenames are assumed to be numbered by chromosome index.
Example: -i example --first_chr 1 --last_chr 2 assumes example_chr1.anc, example_chr1.mut, example_chr2.anc, example_chr2.mut as input. - --chr
- Optional: File containing chromosome names (one per line). Overrides --first_chr and --last_chr.
Example: -i example --chr chr.txt assumes example_chr*.anc, example_chr*.mut as input. - --bins
- Optional: Specify epoch bins. Format: lower, upper, stepsize for function c(0,10^seq(lower, upper, stepsize)).
- --years_per_gen
- Optional: Years per generation. Default: 28.0
PATH_TO_RELATE/bin/RelateCoalescenceRate \
--mode EstimatePopulationSize\
-i example \
-o example Sometimes we want to obtain output with and without specifying the option --poplabels, or we want to specify different poplabels. For this, we can extract coalescence rates from example.bin using
PATH_TO_RELATE/bin/RelateCoalescenceRate \
--mode FinalizePopulationSize\
--poplabels example.poplabels \
-i example \
-o example
Reestimate branch lengths
Once the average mutation rate and coalescence rates have been calculated, we can re-estimate the branch lengths.
- --anc
- Filename of the .anc file
- --mut
- Filename of the .mut file
- -o,--output
- Filename of output files without file extensions.
- -m,--mutation_rate
- Mutation rate per base per generation.
- --mrate
- Specifies the file containing the average mutation rate. Generated using this function.
- --coal
- Specifies the file containing coalescent rates. Generated using this function (do not specify option --poplabels for this).
- --dist
- Optional: Specifies distances between SNPs. If unspecified, we use the distances given in the .mut file. This option is only needed if some SNPs (and trees) have been removed using, e.g., this function. The .dist file can be generated using this function.
- --seed
- Optional: Specifies a seed for the random number generator used in the MCMC algorithm for estimating branch lengths.
PATH_TO_RELATE/bin/RelateCoalescenceRate \
--mode ReEstimateBranchLengths\
--anc example.anc \
--mut example.mut \
--mrate example_avg.rate \
--coal example.coal \
-m 1.25e-8 \
-o example_updated With the updated branch lengths, we can then re-estimate the average mutation and coalescence rates. We can iterate these steps until convergence to obtain an algorithm that simulatneously estimates mutation and coalescence rates and branch lengths.
Estimate mutation rates
Calculate the average mutation rate through time
- -i,--input
- Filename of the .anc and .mut files, excluding the file extensions.
- -o,--output
- Filename of output files without file extensions.
- --first_chr & --last_chr
- Optional: Allow for multiple input files, where filenames are assumed to be numbered by chromosome index.
Example: -i example --first_chr 1 --last_chr 2 assumes example_chr1.anc, example_chr1.mut, example_chr2.anc, example_chr2.mut as input. - --chr
- Optional: File containing chromosome names (one per line). Overrides --first_chr and --last_chr.
Example: -i example --chr chr.txt assumes example_chr*.anc, example_chr*.mut as input. - --bins
- Optional: Specify epoch bins. Format: lower, upper, stepsize for function c(0,10^seq(lower, upper, stepsize)).
- --years_per_gen
- Optional: Years per generation. Default: 28.0
- --dist
- Optional: Specifies .dist file. The .dist file stores distances in bp between SNPs. If unspecified, we use the distances given in the .mut file. This option is only needed if some SNPs (and trees) have been removed using, e.g., this function. The .dist file can be generated using this function. If specified together with --first_chr, --last_chr, please specify the filename without file extension.
PATH_TO_RELATE/bin/RelateMutationRate \
--mode Avg\
-i example \
-o example Calculate the mutation rates grouped by nucleotide in sequence
It is necessary to have appended SNP annotation with --annot to use this function (This can also be done after inferring .anc,.mut files, see ).
- -i,--input
- Filename of the .anc and .mut files, excluding the file extensions.
- -o,--output
- Filename of output files without file extensions.
- --ancestor
- Fasta file containing ancestral genome. This is case insensitive.
- --mask
- Fasta file of same length as the ancestral genome contining a genomic mask. Loci passing the mask are denoted by P, loci not passing the mask are denoted by N. This is case insensitive.
- --bins
- Optional: Specify epoch bins. Format: lower, upper, stepsize for function c(0,10^seq(lower, upper, stepsize)).
- --years_per_gen
- Optional: Years per generation. Default: 28.0
- --dist
- Optional: Specifies .dist file. The .dist file stores distances in bp between SNPs. If unspecified, we use the distances given in the .mut file. This option is only needed if some SNPs (and trees) have been removed using, e.g., this function. The .dist file can be generated using this function. If specified together with --first_chr, --last_chr, please specify the filename without file extension.
- --first_chr & --last_chr
- Currently not supported for mode WithContextForChromosome (because we need to specify separate mask, ancestor files for each chromosome.)
You can however apply mode WithContextForChromosome to each chromosome individually. You can then use mode Finalize (see below) with options --first_chr and --last_chr to obtain a genome-wide estimate. Filenames are assumed to be indexed by chromosome as before.
Example: -i example --first_chr 1 --last_chr 2 assumes example_chr1.anc, example_chr1.mut, example_chr2.anc, example_chr2.mut as input. - --chr
- Optional: File containing chromosome names (one per line). Overrides --first_chr and --last_chr.
Example: -i example --chr chr.txt assumes example_chr*.anc, example_chr*.mut as input.
PATH_TO_RELATE/bin/RelateMutationRate \
--mode WithContextForChromosome \
--mask genomic_mask.fa \
--ancestor ancestor.fa \
-i example_chr1 \
-o example_chr1 Next, to extract the rates from example.bin, use
PATH_TO_RELATE/bin/RelateMutationRate \
--mode Finalize \
-i example_chr1 \
-o example_chr1 If you applied mode WithContextForChromosome to multiple chromosome, use the following to obtain a .bin file summarizing estimates genome-wide. Input filenames are assumed to be example_chr1.bin, example_chr2.bin, etc.
PATH_TO_RELATE/bin/RelateMutationRate \
--mode Finalize \
--first_chr 1 \
--last_chr 22 \
-i example \
-o example Detect positive selection
Detect selection
Calculates p-values for selection evidence from genealogy.
We recommend that you first estimate the population size history using this module, with setting --threshold 0. This is so that the branch lengths in all trees are updated for the estimated population size history. Alternatively, if the region you are analysing is short, you can specify a .coal file in this module and the script reestimates branch lengths for the trees of interest before calculating selection p-values.
P-values can only be calculated for mutations with derived allele frequency of at least 3. Mutations with a smaller DAF are filtered out if the DAFs are appended to the .mut file using RelateFileFormats --mode GenerateSNPAnnotations, otherwise we set the -log10 p-value column to 1.
- -i,--input
- Filename of .anc and .mut files without file extension. Needs to contain all SNPs in haps/sample input file.
- -o,--output
- File of the output files without file extensions.
- -m,--mu
- Mutation rate, e.g., 1.25e-8.
- --first_bp
- Optional: First bp position.
- --last_bp
- Optional: Last bp position.
- --years_per_gen
- Optional: Years per generation. Default: 28.0
- --coal
- Optional: Estimate of coalescent rate, treating all haplotypes as one population. Not necessary if .anc/.mut files were obtained using EstimatePopulationSize.sh, otherwise strongly recommended.
- --seed
- Optional: Random seed for branch lengths estimation. Only used if --coal is specified.
PATH_TO_RELATE/scripts/DetectSelection/DetectSelection.sh \
-i example \
-o example_selection \
-m 1.25e-8 \
--first_bp 1000000 \
--last_bp 1000000 \
--years_per_gen 28 \
--coal popsize.coal
File formats
More details
Below, we describe details about the functions used to calculate the selection p-value.Get selection pvalues
We first calculate, for each SNP, its frequency through time. This information is stored in two files: the .freq file and the .lin file. The former stores the frequency of a SNP through time, the latter stores the number of lineages in the corresponding tree through time.
PATH_TO_RELATE/bin/RelateSelection \
--mode Frequency \
-i example \
-o example We then use these two files to calculate a p-value for selection evidence for each SNP.
PATH_TO_RELATE/bin/RelateSelection \
--mode Selection \
-i example \
-o example Sample branch lengths
Samples branch lengths using MCMC given a coal file.
The output file format is either anc/mut (with multiple branch lengths stored in the anc file), or input file formats of CLUES or PALM.
The output file format for CLUES resembles that of ARGweaver, which is the input file format required by CLUES for inference of selection and inferring allele frequency trajectories.
In Relate, we map mutations to branches with approximately the same descendants as carriers of the derived allele to be robust to errors. Therefore, we output an additional .sites file in which only the descendants of the branch to which the mutation is mapped are marked as derived, the rest are ancestral. Please also note that some SNPs don't map to a unique branch in the tree and such SNPs are left out of this .sites file.
- -i,--input
- Filename of .anc and .mut files without file extension. Needs to contain all SNPs in input haps/sample files. If not, see --dist option.
- -o,--output
- File of the output files without file extensions.
- -m,--mu
- Mutation rate, e.g., 1.25e-8.
- --coal
- Estimated coalescent rate, treating all haplotypes as one population.
- --format
- Output file format. 'a': anc/mut, 'n': newick/sites (used by CLUES), 'b': binary (used by CLUES and PALM). Default: 'a'. Note that anc/mut is modified to store multiple branch lengths for any one branch.
- --num_samples
- Number of times branch lengths are sampled. Integer >= 1.
- --num_proposals
- Optional: Number of MCMC proposals between samples. Default is max(10*N, 1000), where N is the number of haplotypes. If num_proposals=0, all samples will be identical to the input.
- --first_bp
- Optional: First bp position.
- --last_bp
- Optional: Last bp position.
- --dist
- Optional: Filename containing bp positions of SNPs and distances between adjacent SNPs (file format). E.g., obtained using RelateExtract --mode AncMutForSubregion. Required if anc/mut file only covers a subregion of input haps/sample files.
- --seed
- Optional: Random seed for branch lengths estimation.
PATH_TO_RELATE/scripts/SampleBranchLengths/SampleBranchLengths.sh \
-i example \
-o example_sub \
-m 1.25e-8 \
--coal popsize.coal \
--format a \
--num_samples 5 \
--first_bp 100000 \
--last_bp 200000 \
--seed 1
File formats
Plot trees of interest
TreeView
Plots a tree of interest using R.
Requires libraries ggplot2, cowplot.
- --anc
- Filename of the .anc file.
- --mut
- Filename of the .mut file.
- --haps
- Filename of haps file.
- --sample
- Filename of sample file.
- --poplabels
- Filename of a .poplabels file. The file format is described here. This file needs to always contain all samples.
- --bp_of_interest
- Integer specfifying the BP position of the SNP of interest
- --years_per_gen
- Years per generation. Default: 28.0
- -o,--output
- File of the output files without file extensions.
PATH_TO_RELATE/scripts/TreeView/TreeView.sh \
--haps data/example.haps \
--sample data/example.sample \
--anc example.anc \
--mut example.mut \
--poplabels data/example.poplabels \
--bp_of_interest 3000000 \
--years_per_gen 28 \
-o example You can change plotting parameters by editing PATH_TO_RELATE/scripts/TreeView/treeview.R. Specifically, lines 9 - 22 directly influence the plotting parameters.
TreeViewMutation
Plots a tree of interest using R and highlights lineages carrying derived allele.
Requires libraries ggplot2, cowplot.
Takes same arguments as the TreeView.sh script, however bp_of_interest has to specify a SNP in the input.
PATH_TO_RELATE/scripts/TreeView/TreeViewMutation.sh \
--haps data/example.haps
--sample data/example.sample \
--anc example.anc \
--mut example.mut \
--poplabels data/example.poplabels \
--bp_of_interest 3000000 \
--years_per_gen 28 \
-o example TreeViewSamples
Plots a tree of interest using R for an anc/mut file containing multiple sampled branch length (obtained using the SampleBranchLengths module).
Requires libraries ggplot2, cowplot.
Takes same arguments as the TreeView.sh script.
PATH_TO_RELATE/scripts/TreeView/TreeViewSamples.sh \
--haps data/example.haps
--sample data/example.sample \
--anc example.anc \
--mut example.mut \
--poplabels data/example.poplabels \
--bp_of_interest 3000000 \
--years_per_gen 28 \
-o example Extract subtrees, trees in region of interest, and trees in newick format.
Extract subtrees corresponding to a subpopulation
Extracts subtrees such that all tips belong to the populations of interest.
- --anc
- Filename of the .anc file.
- --mut
- Filename of the .mut file.
- --poplabels
- Filename of a .poplabels file. The file format is described here. This file needs to always contain all samples.
- --pop_of_interest
- Specifies populations for which population size is estimated. The second column of the .poplabels file is used for population labels. You can specify multiple populations separated by commas, e.g. pop1,pop2.
- -o,--output
- File of the output files without file extensions.
PATH_TO_RELATE/bin/RelateExtract\
--mode SubTreesForSubpopulation\
--anc example.anc \
--mut example.mut \
--poplabels example.poplabels \
--pop_of_interest group1 \
-o example_subpop Extract trees corresponding to a subregion
Extracts trees in region of interest.
Please always apply this on the anc/mut files inferred using Relate (from haps/sample files), i.e., do not apply repeatedly to get "subregions of subregions". If you do, ignore the output .dist file and use the .dist file obtained from the original anc/mut files instead.
- --anc
- Filename of the .anc file.
- --mut
- Filename of the .mut file.
- --first_bp
- BP of start of region.
- --last_bp
- BP of end of region.
- -o,--output
- File of the output files without file extensions.
PATH_TO_RELATE/bin/RelateExtract\
--mode AncMutForSubregion\
--anc example.anc \
--mut example.mut \
--first_bp 0 \
--last_bp 10000 \
-o example_subregion Extract tree in region of interest in newick format
Extracts trees in region of interest in newick format.
- --anc
- Filename of the .anc file.
- --mut
- Filename of the .mut file.
- -o,--output
- File of the output files without file extensions.
- --bp_of_interest
- Integer specfifying the BP position of interest
- --first_bp
- Integer specfifying the first BP position of interest
- --last_bp
- Integer specfifying the last BP position of interest
PATH_TO_RELATE/bin/RelateExtract\
--mode AncToNewick \
--anc example.anc \
--mut example.mut \
--first_bp 3000000 \
--last_bp 4000000 \
-o example Remove trees with few mutations
Removes trees and associated SNPs from the .anc/.mut files onto which less than a given number of SNPs are mapped.
- --anc
- Filename of the .anc file.
- --mut
- Filename of the .mut file.
- --threshold
- Float in [0,1] which is the fraction of trees to be dropped, ranked by number of mutations mapping. This option serves two purposes: First, it removes bad/uninformative trees. Second it speeds up the inference. As we increase this value, we remove more trees. Default: 0.5.
- -o,--output
- File of the output files without file extensions.
PATH_TO_RELATE/bin/RelateExtract\
--mode RemoveTreesWithFewMutations \
--anc example.anc \
--mut example.mut \
--threshold 0.5 \
-o example_subset Extract .dist file from .mut file.
Extracts distances between SNPs from the .mut file (3rd and 4th column). The .dist file is needed in some applications, when the .anc/.mut files have been subsetted using, e.g., this function In these cases, the .dist file needs to be generated using the not subsetted .mut file.
- --mut
- Filename of the .mut file.
- -o,--output
- File of the output files without file extensions.
PATH_TO_RELATE/bin/RelateExtract\
--mode ExtractDistFromMut \
--mut example.mut \
-o example Divide the .anc/.mut files into smaller files to allow multi-threading.
Dividing the .anc/.mut files into smaller files to allow for multi-threading.
- threads
- Number of threads that will be used. This slightly varies the number of chunks into which the .anc/.mut files will get divided into.
- --anc
- Filename of the .anc file.
- --mut
- Filename of the .mut file.
- -o,--output
- File of the output files without file extensions.
PATH_TO_RELATE/bin/RelateExtract\
--mode DivideAncMut \
--threads 5 \
--anc example.anc \
--mut example.mut \
-o example Combine the .anc/.mut files into one file.
Combining the .anc/.mut files into one file. This should be used after dividing .anc/.mut files using this function.
- -o,--output
- File of the output files without file extensions.
In addition, we assume that the corresponding .param file is in the same directory. You can also manually create this file, which has the header "NUM_HAPLOTYPES NUM_SNPS NUM_TREES NUM_CHUNKS", followed by the corresponding entries on the next line.
PATH_TO_RELATE/bin/RelateExtract\
--mode CombineAncMut \
-o example